![]()
![]()
![]()
Basic Properties of Fluorescence Spectra
Absorption of light
Absorption of light by a molecule causes the excitation of an electron, and the electron moves from a ground state to an excited state. Each of these electronic states may contain a number of vibrational levels. Absorption of light is from the lowest electronic/vibrational state to a number of vibrational levels in the excited electronic state. The different energies of these transitions results in fine structure of the absorption spectra. This fine structure is often not seen in the absorption spectra of chromophores in solution because of inherent line broadening or due to instrumental artifacts (i.e. slit width too large).
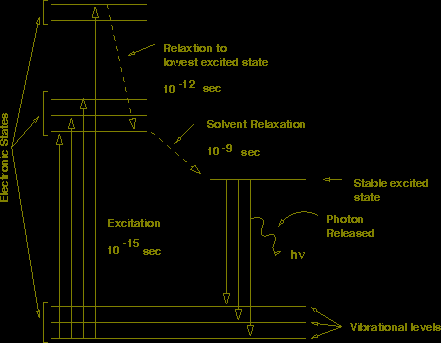
FIGURE 1: Energy Levels of the Excited Electronic State
After the electron has been excited it rapidly relaxes from the higher vibrational states to the lowest vibrational state of the excited electronic state. The rate for this relaxation is on the order of picoseconds and usually occurs before measurements can be made on the system. After reaching the lowest vibrational state of the excited electronic state the excited state can decay to the ground state by a number of mechanisms. The system can lose the energy by internal conversion (heat), quenching (external conversion), by emission of a photon (fluorescence), or by intersystem crossing (phosphorescence). Inter-system crossing produces a triplet state, where the spins of the excited and ground state electrons are no longer paired. Since the emission from the triplet state is forbidden, this state is very stable and can have lifetimes (msec to sec). Most compounds decay by non-radiative processes (such as heat) and are therefore not fluorescent. Fluorescent compounds, on the other hand, decay to the ground state by the emission of light.
The energy of the photon that is emitted as the electron decays to the ground state depends on the energy difference between the excited and ground state at the time of emission . The rapid decay of excited vibrational states generally implies that the state from which the system decays is independent of the excitation wavelength. However, the state to which the system decays is not necessarily the lowest vibrational state of the ground electronic state. Therefore, the emission spectra of fluorescent compounds will also show fine structure.
In theory, the energy of the transition from the lowest energy states is the same for both absorption and emission. In practice, the average energy of the emitted photon is generally less than the corresponding absorption band. This red shift is due to a change in the local environment of the excited state during it's lifetime. The re-organization of solvent dipoles will lower the energy of the excited state, causing a red shift in the emission spectra. This shift in the emission spectrum is called a Stokes shift. The magnitude of the Stokes shift depends on the polarity of the solvent. Usually, solvents of higher polarity produce larger Stokes shifts. Note that shifts of this type are also seen in absorption spectroscopy, but in that case they are a reflection of stabilization of the empty excited state orbital.
Spontaneous Emission and Quantum Yield
The rate of light absorption by a compound is equal to the Einstein B co-efficient:
![]() (1)
(1)
This is also equal to the rate of stimulated emission from the excited
state to the ground state because the transition dipole operator,![]() , is the same for both excitation and emission. The process of returning
from the excited state to the ground state with the emission of a photon
in phase with the exciting electromagnetic field is referred to as stimulated
emission .
, is the same for both excitation and emission. The process of returning
from the excited state to the ground state with the emission of a photon
in phase with the exciting electromagnetic field is referred to as stimulated
emission .
If transitions in optical systems were caused by absorption and stimulated emission then we would expect the equilibrium populations of the two states to be equal since the rate constants are equal. We know that this is not the case for visible spectroscopy, thus there must be other mechanisms for the excited state to return to the ground state. One mechanism is spontaneous emission of the excited atom. In contrast to stimulated absorption and emission, spontaneous emission does not require the existence of an electromagnetic field at the resonance frequency. The rate of spontaneous emission is given by the Einstein A coefficient. Spontaneous emission from the excited state is just one of the mechanism which limits the lifetime of the excited state. A is typically quite large for excited states that emit at ultraviolet or visible wavelengths (i.e. A greater than 109 sec-1).
The rate of spontaneous emission is the rate at which the fluorescent molecule will emit light. However, the amount of fluorescence obtained from a molecule depends on the rate of spontaneous emission versus other mechanisms which return the electron to the ground state. Stimulated emission is not significant at most fluences of light used in the laboratory and need not be considered further. If other mechanisms for relaxation to the ground state besides spontaneous emission exist then the lifetime of the excited state is given by:
![]() (2)
(2)
Where ko = 1 /A, kq[Q] is the relaxation rate due to quenching and kother includes any relaxation processes besides spontaneous emission or quenching.
This lifetime is the observed fluorescent life time because relaxation of the system to the ground state by any mechanism results in a loss of excited state molecules. The quantum yield is the ratio of the rate constant for fluorescent decay versus the sum of all pathways:
![]() (3)
(3)
The quantum yield is simply the probability that an excited system will return to the ground state by the emission of a photon. For a steady state experiment the quantum yield is the number of photons emitted/photons absorbed.
The quantum yield is also given by the ratio of lifetimes (this follows directly from the eq. above:
![]() (4)
(4)
Fluorescent Molecules
Intrinsic Fluorescent Molecules: The quantum yield of a fluorophore is a function of the molecular structure. Of the common groups found in biological macromolecules (and their assemblies) only the following have significant fluorescence (high enough quantum yield) to be of much use in fluorescence spectroscopy:
Note that all of these molecules consist of planar aromatic rings.
Since the number of fluorescent molecules in a experimental system is small and it is often possible to observe fluorescence from a single group within a macromolecule. Therefore fluorescent spectroscopy is capable of providing information on individual groups within macromolecular structures.
Extrinsic Fluorescent Molecules (Probes): Often it is necessary to add an external molecule which is fluorescent (a fluorescent probe) to the system of interest. Labeling with a fluorescent probe may be necessary to obtain fluorescence from samples that have little or no intrinsic fluorescence (i.e. lipids). Labeling is also performed when it is desirable to introduce a fluorescent molecule with unique properties into the system.
There are two type of extrinsic fluorescent molecules, covalent and non-covalent. An example of a covalent probe is fluorescein 5'-isothiocyanate which can be attached to lys residues in proteins. An example of a non-covalent probe is perylene, which could be used to investigate the properties of lipid bilayers.
Application of Fluorescence Spectroscopy
Various characteristics of fluorescence spectroscopy make this technique suitable for many applications in biochemistry. One important characteristic is the sensitivity of fluorescence spectroscopy. The source of the sensitivity difference between absorbance and fluorescence spectroscopy is quite clear. In absorbance spectroscopy the PMT must be able to detect a slight difference in the number of photons which pass through the sample and reference cell. In the case of fluorescence only the emitted photons are counted and it is possible to detect on concentrations in the order of nM. Further, since few "natural" molecules are fluorescent it is easier to determine where your signals are coming from. The down side of this is that you can only gain information for those parts of the system that are fluorescent.
Another important characteristic of fluorescence spectroscopy is the sensitivity of the fluorophore to the environment. The fluorescence lifetime, quantum yield, and emission spectra of a fluorophore can be sensitive to the local environment. The effect of the environment on the excited state is much more pronounced with fluorescence spectroscopy than with absorption spectroscopy because of the additional time available to sample/modify the environment of the excited chromophore. These changes in the properties of fluorescence make fluorescence spectroscopy a widely used method of detecting a change in the environment of the fluorophore. A common and useful example is ethidium bromide. This compound is not fluorescent in water but becomes fluorescent in the presence of DNA. The fluorescence of the ethidium bromide is said to be quenched in water. The quenching is due to the interaction of the excited ethidium molecules with a polar solvent, leading to non-radiative decay of the excited state. A number of compounds have been developed with fluorescent properties that change when the compound interacts with Ca2+. These compounds provide a non-destructive way to measure the intra-cellular Ca2+ levels.
Finally, the coupling between the transition dipoles of fluorescent molecules provides a mechanism to measure the distance between these groups on larger macromolecules. The distances that can be measured are impressive, on the order of 20-70A, making fluorescence spectroscopy unique in this application.
Fluorescence Quenching
The definition of quenching is any process which reduces the lifetime of the excited state. A reduction in the lifetime usually implies a decrease in the quantum yield:
![]() (5)
(5)
Some of the processes which reduce the quantum yield are:
Note that light scattering can appear as quenching (loss of fluorescent signal).
Collisional and static quenching require contact between the fluorophore and the quenching. Thus, these methods are useful to measure rates of diffusion and exposure of fluorescent species to the quencher. A large number of quenchers are known and a partial list is:
molecular oxygen, amides, BrO4-, xenon, I-, peroxides, nitroxides, acrylamide
The mechanism of quenching has been difficult to determine. In some cases, such as acrylamide, the quenching mechanism appears to involve a transfer of charge from the excited fluorophore to the quenching agent.
Theory of Collisional Quenching
It is experimentally observed that quenchers reduce the fluorescent intensity in a manner proportional to the concentration of the quencher. This is summarized by the Stern-Volmer equation:
![]() (6)
(6)
Fo is the intensity in the absence of Q
F is the intensity in the presence of Q
![]() is the lifetime in the absence
of Q
is the lifetime in the absence
of Q
kq is the quenching constant
The derivation of this equation follows.
The measured fluorescence is proportional to the quantum yield:
![]() (7)
(7)
![]() (8)
(8)
The quantum yield in the presence of the quencher is:
![]() (9)
(9)
The ratio of the fluorescence intensities in the absence and presence of the quencher are:
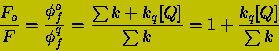 (10)
(10)
![]() (11)
(11)
The "traditional" method of obtaining the quenching constant
![]() is to make a Stern-Volmer
plot, which is a plot of
is to make a Stern-Volmer
plot, which is a plot of ![]() versus
[Q]. The slope of the line gives
versus
[Q]. The slope of the line gives ![]() .
However, linearization of the data by this method results in severe distortion
of the error associated with the data. Therefore, if you are doing this
for a living, it is better to fit the data to the non-linear equation:
.
However, linearization of the data by this method results in severe distortion
of the error associated with the data. Therefore, if you are doing this
for a living, it is better to fit the data to the non-linear equation:
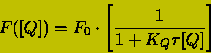 (12)
(12)
Collisional quenching requires the fluorophore and the quencher to bump into each other while the fluorophore is in the excited state, resulting in quenching. The rate constant for collisional quenching is given by:
![]() (13)
(13)
![]() is the quenching
efficiency
is the quenching
efficiency
kc is the bimolecular collision rate.
The bimolecular collision rate can be estimated from the diffusion constants of both the fluorophore and the quencher:
![]() (14)
(14)
rf , rq are the molecular radii of the fluorophore and the quencher in centimeters. Df , Dq are the translational diffusion coefficients, N is Avogadro's' number N/1000 converts from molarity to molecules/cm3
The most common use of quenching in the study of proteins is to determine the location of Trp residues, or the change in location of Trp residues, due to conformational changes. In general, a reduction in the quenching of Trp residues bound to proteins versus that of free Trp is observed, and this can arise from two factors. First, the quenching of Trp is reduced to that of free Trp (or indole) because the Trp is now attached to a molecule with a smaller diffusion co-efficient. For proteins greater than 50 KDa in size, this results in a decrease in the quenching constant of 50 The second factor arises from the fact that the Trp residue is buried in the protein and is thus not accessible to quenching agent unless the quenching agent can diffuse into the protein interior. The correlation between acrylamide quenching and the emission wavelength of the Trp residue suggests that Trp residues buried in hydrophobic regions of the protein are less accessible for quenching.
In general, the quenching of Trp fluorescence by I- or acrylamide can be regarded as being entirely due to collisional quenching. In many cases the amount of quenching due to I- versus that observed by acrylamide can be quite different. This appears to be related to the local environment of the Trp residue. If the Trp residue is found in a region of high negative charge density it is likely that quenching due to I- will smaller than the quenching observed by with acrylamide.
Multi-Fluorophore Proteins: Often, proteins contain more than one fluorophore, and each fluorophore can be quenched to different levels by the quenching agent. In this case the quenching of the fluorescence is described by the following equation:
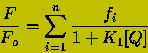 (15)
(15)
where fi and Ki are the fractional contribution to the fluorescence of the ith species and the quenching constant for the ith species, respectively.
Static Quenching
Static quenching is observed when a fluorophore forms a complex with the quencher. It is normally assumed that complex formation will prevent the fluorophore from emitting a fluorescent photon, however those fluorophores which are not complexed with the quencher emit normally with the same lifetime as in the absence of the quencher. The reduction in fluorescence can be calculated from the binding constant of the following reaction scheme:
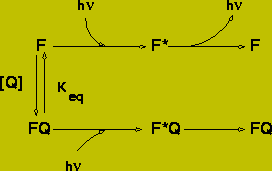 (16)
(16)
The fluorescent intensity ratio is:
![]() (17)
(17)
![]() (18)
(18)
= 1 + Keq [Q]
![]() (19)
(19)
Note: The value for [Q] is that for the concentration of unbound quencher.
Static quenching is a very good method of measuring binding affinities (if the ligand quenches) because very little material is required. A potential problem with measurements of this type is the quenching of the fluorophore by the unbound quencher, principally through the effects of non-radiative energy transfer. This is only a serious problem for low-affinity binding sites.
Non-linear Stern-Volmer plots will arise if either the quenching of the fluorophore by the bound quencher is not complete, or if additional fluorophores are present in the protein which are not quenched when ligand binds.
Note that the relationship between Fo/F and [Q] is the same for both collisional and static quenching. These can only be distinguished by lifetime measurements:
![]() (static)
(static)
![]() (collisional)
(collisional)
Fluorescence Energy Transfer
Energy transfer is the non-radiative transfer of energy from a donor to an acceptor. Since the rate of transfer depends on the distance between the donor and acceptor, energy transfer can be used to measure distances between sites on biopolymers. The distance range is much larger (20-70A) than possible with other spectroscopic techniques (e.g. NMR - 5 A), thus making fluorescence energy transfer a very useful technique.
For energy transfer to occur the absorption spectra of the acceptor must overlap the emission spectra of the donor. This should not be confused with the trivial "inner-filter" effect (absorption of the emitted light by the acceptor). Instead the overlap is required such that quantum energy levels of equivalent energy exist for productive dipolar coupling between the two molecules.
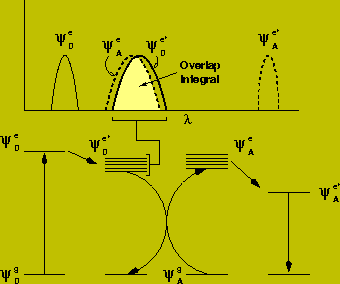
The effect of this transfer is to reduce the lifetime of the excited state of the donor:
![]() (20)
(20)
Where:
kD is the fluorescence decay time of the donor
koD is the spontaneous emission rate of the donor
kotherD refer to other mechanism of decay, such as quenching
kT is the transfer rate from donor to acceptor
The efficiency of transfer is given by:
![]() (21)
(21)
Alternate expressions are:
![]() (22)
(22)
![]() is the lifetime
of the donor when the acceptor are present
is the lifetime
of the donor when the acceptor are present
![]() is the lifetime
of the donor in the absence of the acceptor
is the lifetime
of the donor in the absence of the acceptor
The quantum yield of the donor fluorescence, in the absence of the acceptor, is given by:
![]() (23)
(23)
Where koD is the rate of spontaneous emission of the donor in the absence of the acceptor. This is not likely to be affected by the presence of the acceptor, thus the efficiency of transfer is also related to the fluorescent intensity:
![]() (24)
(24)
The actual rate of transfer is calculated by considering a dipolar interaction between the two chromophores.
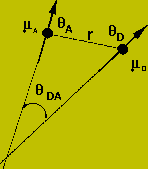
This rate will be proportional to the square of the dipole operator:
![]() (25)
(25)
The dipolar Hamiltonian (coupling energy) is:
![]() (26)
(26)
Note that this is exactly the same equation that was discussed with respect to dipole coupling in optical spectroscopy.
Then end result of a lot of math is the following equation:
![]() (27)
(27)
![]() (28)
(28)
![]() is the fraction
of donors which emit light at frequency
is the fraction
of donors which emit light at frequency ![]() .
.
The rate of transfer is composed of four parts:
1.
![]() (29)
(29)
contains information on the orientation and distance between the donor and acceptor.
2.
![]() (30)
(30)
represents the strength of the donor transition dipole.
3.
![]() (31)
(31)
represents the strength of the acceptor transition dipole.
4.
![]() (32)
(32)
represents the fraction of donor molecules which emit light that is within the absorption band of the acceptor.
Equation 27 can be written as:
![]() (33)
(33)
Where the distance, Ro, is called the Forster distance. Ro is the distance between donor and acceptor at which the energy transfer efficiency is 0.5.
Equation 33 leads to another form for the transfer efficiency:
![]() (34)
(34)
Ro can be calculated using the following expression:
![]() (35)
(35)
![]() (36)
(36)
The 1/r6 dependence was verified by Stryer. They determined the efficiency of transfer, E, for different values of R and plotted:
![]() (37)
(37)
They found j=5.9 +/- 0.3. They also measured the polarization of the
acceptor fluorescence and found it to be zero (i.e. ![]() =
2/3 ).
=
2/3 ).
Limitations of Energy Transfer: The energy transfer efficiency depends
on the orientation factor, the refractive index, the quantum yield of the
donor, and the overlap integral between the emission spectrum of the donor
and the absorption spectra of the acceptor. All of these factors affect
the energy transfer, and thus the measurement of distances between donor
and acceptor. The orientation factor,![]() , is the source of most of the error in measurements of distances with
energy transfer.
, is the source of most of the error in measurements of distances with
energy transfer. ![]() varies
between 0 and 4, depending on the orientation of the fluorophores. It is
generally assumed that the orientation of donor acceptor pairs is random,
thus the average value of
varies
between 0 and 4, depending on the orientation of the fluorophores. It is
generally assumed that the orientation of donor acceptor pairs is random,
thus the average value of ![]() is
2/3.
is
2/3.
Application of Energy Transfer
Measurement of Binding - This is commonly used to study ligand binding to proteins. In this case the Trp fluorescence is transferred to the bound ligand and the transfer efficiency can be used to determine the occupancy of the binding site. Note that the ligand need not be fluorescent as long as the absorbance spectra of the ligand overlaps with the Trp emission spectra transfer will occur. This technique is easy to apply to systems with high binding affinities. In the case when binding affinities are low it is necessary to correct for the transfer of fluorescence to unbound ligand.
Distance Measurements - Distances can be measured using equation. Although there are a number of assumptions involved with these measurements, the fact that most of the errors are attenuated by the sixth root implies that the overall accuracy is quite good. The only exception occurs when the donor/acceptor pairs are restricted in motion, resulting in a non-averaged value for . The measurable distance can be varied by altering the donor/acceptor pair.
In many cases a single distance cannot account for the measured energy transfer. This is most obvious is time resolved energy transfer because the decay of the fluorescence in the presence of the acceptor is not a single exponential. The actual decay will depend on the probability distribution, P(r), of the donor and acceptor pairs to give the following decay rate for the fluorescence:
![]() (38)
(38)
Association between Proteins - The association between proteins can be investigated by labeling on species with a donor and one with an acceptor. The presence of energy transfer is indicative of complex formation. Note that both species can be the same protein.
Distribution of Fluorophores - The energy transfer between a distribution of donor and acceptors (e.g. labeled lipid molecules) can be used, in conjunction with modeling, to provide information on the distribution of molecules. An example is phase separation of lipid species.